




MPS I
MPS II
MPS III
MPS IVA/ Morquio A
MPS VI
MPS VII
MPS IX
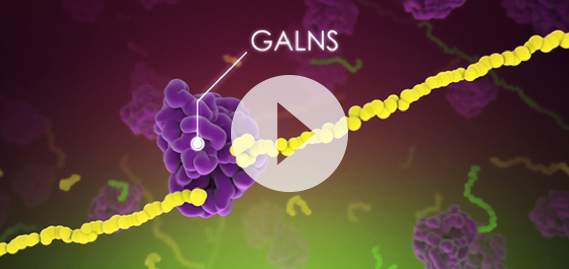 –>
–>Table of MPS disorders and enzymes
–>
Morquio A MOD Video
MPS VI MOD Video
VIMIZIM® (elosulfase alfa) is indicated for patients with Mucopolysaccharidosis type IVA (MPS IVA; Morquio A syndrome).
Life-threatening anaphylactic reactions have occurred in some patients during VIMIZIM® (elosulfase alfa) infusions. Anaphylaxis, presenting as cough, erythema, throat tightness, urticaria, flushing, cyanosis, hypotension, rash, dyspnoea, chest discomfort and gastrointestinal symptoms (e.g. nausea, abdominal pain, retching and vomiting) in conjunction with urticaria, have been reported to occur during VIMIZIM infusions, regardless of duration of the course of treatment. Closely observe patients during and after VIMIZIM administration and be prepared to manage anaphylaxis. Inform patients of the signs and symptoms of anaphylaxis and have them seek immediate medical care should symptoms occur. Patients with acute respiratory illness may be at risk of serious acute exacerbation of their respiratory compromise due to hypersensitivity reactions, and require additional monitoring.
Due to the potential for anaphylaxis, appropriate medical support should be readily available when VIMIZIM is administered and for an appropriate period of time following administration. In clinical trials, cases of anaphylaxis occurred as early as 30 minutes from the start of infusion and up to three hours after infusion, and as late into treatment as the 47th infusion.
In clinical trials, hypersensitivity reactions have been observed as early as 30 minutes from the start of infusion but as late as six days after infusion. Frequent symptoms of hypersensitivity reactions (occurring in more than 2 patients) included anaphylactic reactions, urticaria, peripheral oedema, cough, dyspnoea and flushing.
Because of the potential for hypersensitivity reactions, administer antihistamines with or without antipyretics prior to infusion. Management of hypersensitivity reactions should be based on the severity of the reaction and include slowing or temporary interruption of the infusion and/or administration of additional antihistamines, antipyretics, and/or corticosteroids for mild reactions. However, if severe hypersensitivity reactions occur, immediately stop the infusion of VIMIZIM and initiate appropriate treatment.
Consider the risks and benefits of re-administering VIMIZIM following a severe reaction.
Patients with acute febrile or respiratory illness at the time of VIMIZIM infusion may be at higher risk of life-threatening complications from hypersensitivity reactions. Careful consideration should be given to the patient’s clinical status prior to administration of VIMIZIM and consider delaying the VIMIZIM infusion.
Sleep apnoea is common in MPS IVA patients. Evaluation of airway patency should be considered prior to initiation of treatment with VIMIZIM. Patients using supplemental oxygen or continuous positive airway pressure (CPAP) during sleep should have these treatments readily available during infusion in the event of an acute reaction, or extreme drowsiness/sleep induced by antihistamine use.
Spinal or cervical cord compression (SCC) is a known and serious complication of MPS IVA and may occur as part of the natural history of the disease. In clinical trials, SCC was observed both in patients receiving VIMIZIM and patients receiving placebo. Patients with MPS IVA should be monitored for signs and symptoms of SCC (including back pain, paralysis of limbs below the level of compression, urinary and faecal incontinence) and given appropriate clinical care.
All patients treated with VIMIZIM 2 mg/kg once per week in the placebo-controlled trial developed anti-drug antibodies. The relationship between the presence of neutralising antibodies and long-term therapeutic response could not be determined.
VIMIZIM should be used during pregnancy only if the potential benefit justifies the potential risk to the foetus. It is not known if VIMIZIM is present in human milk. Exercise caution when administering VIMIZIM to a breastfeeding mother. There is a Morquio A Registry that collects data on pregnant women and breastfeeding mothers with MPS IVA who are treated with VIMIZIM. Contact MARS@BMRN.com for information and enrollment.
Safety and effectiveness in paediatric patients below 5 years of age have not been established.
In clinical trials, the most common adverse reactions (≥10%) occurring during infusion included pyrexia, vomiting, headache, nausea, abdominal pain, chills and fatigue. The acute reactions requiring intervention were managed by either temporarily interrupting or discontinuing infusion, and administering additional antihistamine, antipyretics or corticosteroids.
To report SUSPECTED ADVERSE REACTIONS contact BioMarin Pharmaceutical Inc. at 1-866-906-6100, or FDA at 1-800-FDA-1088 or go to www.fda.gov/medwatch.
Please see full Prescribing Information, including boxed warning.
NAGLAZYME® (galsulfase) is indicated for patients with Mucopolysaccharidosis VI (MPS VI; Maroteaux-Lamy syndrome). NAGLAZYME has been shown to improve walking and stair-climbing capacity.
Life-threatening anaphylactic reactions and severe allergic reactions have been observed in some patients during NAGLAZYME (galsulfase) infusions and up to 24 hours after infusion. If these reactions occur, immediate discontinuation of NAGLAZYME is recommended and appropriate medical treatment should be initiated, which may include resuscitation, epinephrine, administering additional antihistamines, antipyretics or corticosteroids. In patients who have experienced anaphylaxis or other severe allergic reactions during infusion with NAGLAZYME, caution should be exercised upon rechallenge; appropriately trained personnel and equipment for emergency resuscitation (including epinephrine) should be available during infusions.
As with other enzyme replacement therapies, immune-mediated reactions, including membranous glomerulonephritis have been observed. In clinical trials, nearly all patients developed antibodies as a result of treatment with NAGLAZYME; however, the analysis revealed no consistent predictive relationship between total antibody titre, neutralising or IgE antibodies, and infusion-associated reactions, urinary glycosaminoglycan (GAG) levels, or endurance measures.
Caution should be exercised when administering NAGLAZYME to patients susceptible to fluid volume overload because congestive heart failure may result. Consider a decreased total infusion volume and infusion rate when administering NAGLAZYME to these patients.
Consideration to delay NAGLAZYME infusion should be given when treating patients who present with an acute febrile or respiratory illness. Sleep apnoea is common in MPS VI patients and antihistamine pretreatment may increase the risk of apnoeic episodes. Evaluation of airway patency should be considered prior to the initiation of treatment. Patients using supplemental oxygen or continuous positive airway pressure (CPAP) during sleep should have these treatments readily available during infusion in the event of an infusion reaction, or extreme drowsiness/sleep induced by antihistamine use.
Pretreatment with antihistamines with or without antipyretics is recommended prior to the start of infusion to reduce the risk of infusion reactions. If infusion reactions occur, decreasing the infusion rate, temporarily stopping the infusion, or administering additional antihistamines and/or antipyretics is recommended.
During infusion, serious adverse reactions included laryngeal oedema, apnoea, pyrexia, urticaria, respiratory distress, angioedema, and anaphylactoid reaction; severe adverse reactions included urticaria, chest pain, rash, abdominal pain, dyspnoea, apnoea, laryngeal oedema and conjunctivitis. The most common adverse events (=10%) observed in clinical trials in patients treated with NAGLAZYME were rash, pain, urticaria, pyrexia, pruritus, chills, headache, nausea, vomiting, abdominal pain and dyspnoea. The most common adverse reactions requiring interventions are infusion-related reactions.
Spinal/cervical cord compression is a known and serious complication that is expected to occur during the natural course of MPS VI. Signs and symptoms of spinal/cervical cord compression include back pain, paralysis of limbs below the level of compression and urinary or faecal incontinence. Patients should be evaluated for spinal/cervical cord compression prior to initiation of NAGLAZYME to establish a baseline and risk profile. Patients treated with NAGLAZYME should be regularly monitored for the development or progression of spinal/cervical cord compression and be given appropriate clinical care.
To report SUSPECTED ADVERSE REACTIONS contact BioMarin Pharmaceutical Inc. at 1-866-906-6100, or FDA at 1-800-FDA-1088 or go to www.fda.gov/medwatch.
Please see full Prescribing Information.
