Decades of ongoing research and clinical experience have produced a new era in the optimal management of mucopolysaccharidosis (MPS) disorders. This rapidly evolving standard of care for MPS relies on the geneticist at the centre of the coordinated, multidisciplinary care-delivery model and provides doctors unmatched opportunities to change patients’ lives.1-3
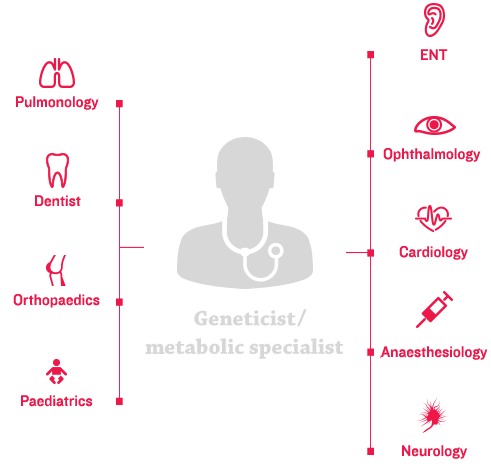
The heterogeneous and variable nature of MPS disorders necessitates a personalised approach to coordinated patient care, starting at the multidisciplinary care-delivery model.4 The aim of coordinated care is to help patients achieve a greater quality of life, which includes:
For paediatric patients with chronic, complex, multisystemic genetic disease such as MPS, care through a coordinated multidisciplinary care-delivery model is associated with reduction in healthcare utilisation and improved health outcomes.5-8
Coordination within the multidisciplinary care-delivery model must be implemented across all elements of the broader healthcare system (e.g. specialty care, hospitals, home healthcare, and community services) and within patients’ individualised management plans.3 Your role in the multidisciplinary care-delivery model can be essential to the implementation of best practices in the management of MPS disorders and improvement of patient outcomes.1-3
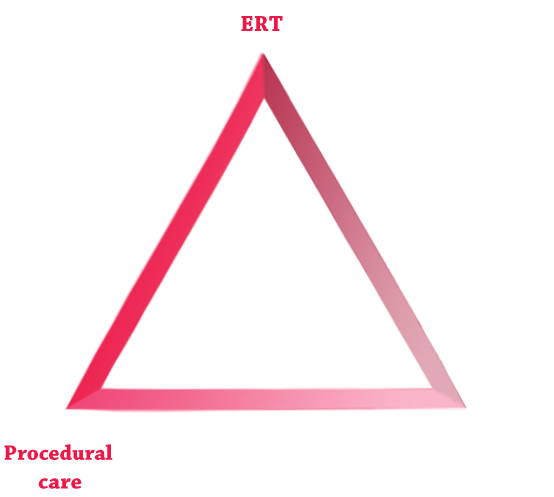
Application of optimal MPS disease management, grouped into the following 3 pillars of care, can help to improve patient outcomes:
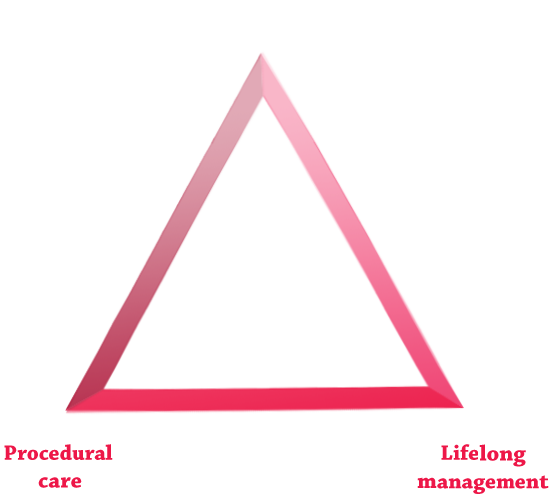
In parallel with a multidisciplinary care-delivery model, orthopaedic specialists play a significant role in shaping individualised management plans that address ERT, lifelong management, and procedural care, ultimately helping to optimise patient outcomes.2,10
ERT, when available, is a cornerstone of therapy.9-11
A patient with rapidly progressing MPS is identified early, enabling prompt initiation of ERT.18

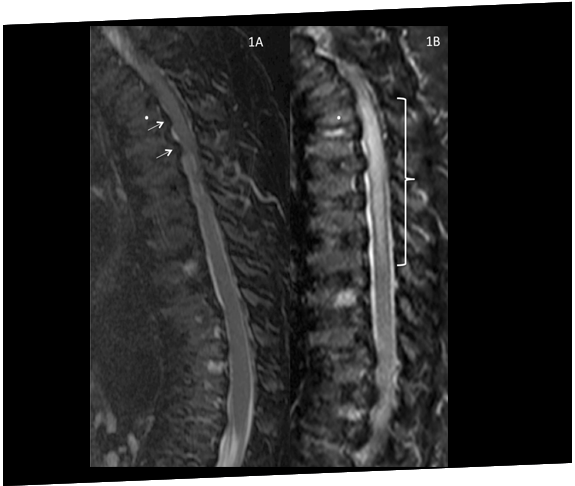
A patient with Morquio A (MPS IVA) develops spinal cord compression and paraplegia immediately after surgery to correct genu valgum, highlighting the issue of vulnerability to compression during anaesthesia.19
A child presenting with perceived developmental delays over a 7-year time period is later diagnosed with MPS VI and started on ERT.18
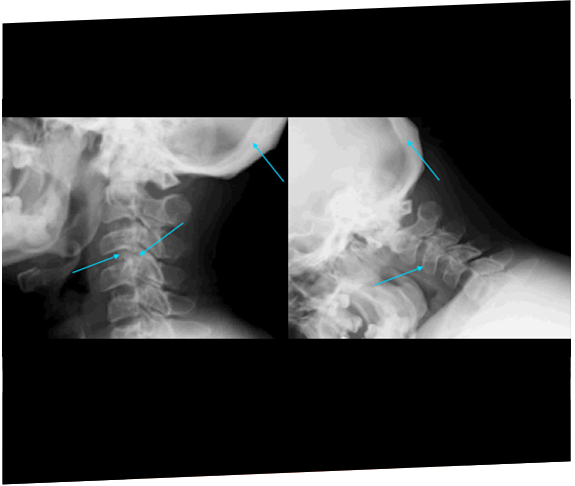
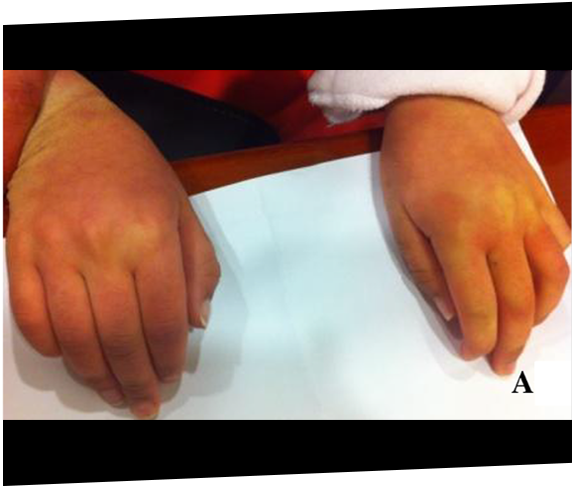
Two siblings were eventually diagnosed with MPS IIIA after 11 years following rapid progression of developmental delay and subsequent referral to a geneticist.20
While each subtype of MPS disorder is clinically distinct, all feature the life-limiting, progressive, multisystemic disease manifestations common to MPS disease pathology.11,13,21,22 Management of patients with MPS requires an understanding of the specific clinical manifestations and management recommendations for each MPS subtype.2,10
November 2015
Optimal bilateral labour analgesia was not achieved despite multiple adjustments, and systemic analgesia was needed for caesarean delivery.
May 2015
Delays to diagnosis occurred due to the lack of or distance to diagnostic facilities, alternative diagnoses and misleading symptoms experienced. Several patients
experienced manifestations that were subtler than would be expected and were subsequently overlooked. Cases also highlighted the unique challenges associated with
diagnosing MPS VI from the perspective of different specialties and provide insights into how these patients initially present.
April 2016
Our current understanding of other cardiac issues in adults with the MPSs, especially with the coronary circulation and myocardium, is meagre and more needs to be
known to effectively care for this emerging population of adults.
References: 1. Hendriksz CJ, Berger KI, Giugliani R, et al. International guidelines for the management and treatment of Morquio A syndrome. Am J Med Genet Part A. 2014;9999A:1-15. doi:10.1002/ajmg.a.36833. 2. Muenzer J. The mucopolysaccharidoses: a heterogeneous group of disorders with variable pediatric presentations. J Pediatr. 2004;144(suppl 5):S27-S34. 3. Agency for Healthcare Research and Quality. Defining the PCMH. https://pcmh.ahrq.gov/page/defining-pcmh. Accessed December 15, 2015. 4. Hendriksz CJ, Harmatz P, Beck M, et al. Review of clinical presentation and diagnosis of mucopolysaccharidosis IVA. Mol Genet Metab. 2013;110:54-64. doi:10.1016/j.ymgme.2013.04.002. 5. Casey PH, Lyle RE, Bird TM, et al. Effect of hospital-based comprehensive care clinic on health costs for Medicaid-insured medically complex children. Arch Pediatr Adolesc Med. 2011;165(5):392-398. doi:10.1001/archpediatrics.2011.5. 6. Mosquera RA, Avritscher EBC, Samuels CL, et al. Effect of an enhanced medical home on serious illness and cost of care among high-risk children with chronic illness: a randomized clinical trial. JAMA. 2014;312(24):2640-2648. doi:10.1001/jama.2014.16419. 7. Klitzner TS, Rabbitt LA, Chang RKR. Benefits of care coordination for children with complex disease: a pilot medical home project in a resident teaching clinic. J Pediatr. 2010;156(6):1006-1010. doi:10.1016/j.jpeds.2009.12.012. 8. Gordon JB, Colby HH, Bartelt T, Jablonski D, Krauthoefer ML, Havens P. A tertiary care-primary care partnership model for medically complex and fragile children and youth with special health care needs. Arch Pediatr Adolesc Med. 2007;161(10):937-944. 9. Hendriksz C. Improved diagnostic procedures in attenuated mucopolysaccharidosis. Br J Hosp Med. 2011;72(2):91-95. 10. Muenzer J, Wraith JE, Clarke LA, International Consensus Panel on the Management and Treatment of Mucopolysaccharidosis I. Mucopolysaccharidosis I: management and treatment guidelines. Pediatrics. 2009;123(1):19-29. doi:10.1542/peds.2008-0416. 11. Muenzer J, Beck M, Eng CM, et al. Long-term, open-labeled extension study of idursulfase in the treatment of Hunter syndrome. Genet Med. 2011;13(2):95-101. doi:10.1097/GIM.0b013e3181fea459. 12. Kakkis ED, Neufeld EF. The mucopolysaccharidoses. In: Berg BO, ed. Principles of child neurology. New York, NY: McGraw-Hill; 1996:1141-1166. 13. Lehman TJA, Miller N, Norquist B, Underhill L, Keutzer J. Diagnosis of the mucopolysaccharidoses. Rheumatology. 2011;50(suppl 5):v41-v48. 14. Lavery C, Hendriksz C. Mortality in patients with Morquio syndrome A. J Inherit Metab Dis Rep. 2015;15:59-66. doi:10.1007/8904_2014_298. 15. Giugliani R, Lampe C, Guffon N, et al. Natural history and galsulfase treatment in mucopolysaccharidosis VI (MPS VI, Maroteaux-Lamy syndrome)—10-year follow-up of patients who previously participated in an MPS VI Survey Study. Am J Med Genet A. 2014;164A(8):1953-1964. doi:10.1002/ajmg.a.36584. 16. Spinello CM, Novello LM, Pitino S, et al. Anesthetic management in mucopolysaccharidoses. ISRN Anesthesiol. 2013;2013:1-10. doi:10.1155/2013/791983. 17. Sam JA, Baluch AR, Niaz RS, Lonadier L, Kaye AD. Mucopolysaccharidoses: anesthetic considerations and clinical manifestations. Middle East J Anaesthesiol. 2011;21(2):243-253. 18. Data on file. Biomarin Pharmaceutical, Inc. 19. Drummond JC, Krane EJ, Tomatsu S, Theroux MC, Lee RR. Paraplegia after epidural-general anesthesia in a Morquio patient with moderate thoracic spinal stenosis. Can J Anesth. 2015;62(1):45-49. doi:10.1007/s12630-014-0247-1. 20. Sharkia R, Mahajnah M, Zalan A, Sourlis C, Bauer P, Schöls L. Sanfilippo type A: new clinical manifestations and neuro-imaging findings in patients from the same family in Israel: a case report. J Med Case Rep. 2014;8:78. doi:10.1186/1752-1947-8-78. 21. Clarke LA, Winchester B, Giugliani R, Tylki-Szymańska A, Amartino H. Biomarkers for the mucopolysaccharidoses: discovery and clinical utility. Mol Genet Metab. 2012;106(4):396-402. doi:10.1016/j.ymgme.2012.05.003. 22. Morishita K, Petty RE. Musculoskeletal manifestations of mucopolysaccharidoses. Rheumatology. 2011;50(suppl 5):v19-v25. doi:10.1093/rheumatology/ker397. 23. Ashworth JL, Biswas S, Wraith E, Lloyd IC. Mucopolysaccharidoses and the eye. Surv Ophthalmol. 2006;51(1):1-17.