


Mucopolysaccharidosis (MPS) VI, or Maroteaux-Lamy syndrome, is a devastating, progressive, and heterogeneous disorder with severe pathologies affecting multiple organs. It is caused by deficient activity of arylsulfatase B (ASB), the enzyme that catabolises the glycosaminoglycans (GAGs) dermatan sulfate and chondroitin sulfate.1
Although there is wide variability in the phenotypic presentation of MPS VI,1,2 patients are often characterised as having either rapidly or slowly progressing disease:
Regardless of rate of progression, untreated MPS VI can progress over years and lead to the following3:
MPS VI is a clinically heterogeneous condition in which patients can present with marked disease in the first year of life or with disease that progresses more slowly, with symptoms presenting over a longer period of time. However, it is important to recognise that MPS VI manifests symptoms along a continuum, so there are no fixed parameters for these categories of disease progression.1,2
Rapidly progressing MPS VI, characterised in the cross-sectional survey study by Swiedler et al as urinary levels of GAG protein (uGAG) above 200 μg/mg,3 can manifest in the first year of life with non-specific symptoms. Between years 2 and 3, characteristic features develop, including the following1,2,5:
Slowly progressing MPS VI, characterised as uGAG levels less than or equal to 200 μg/mg, may not overtly present, thus delaying diagnosis until later in life. Nevertheless, patients develop significant morbidity that may be life limiting and life threatening. Whether rapidly or slowly progressing, MPS VI does not typically cause neurocognitive deficits, although physical limitations can affect patients’ learning and development.3
The table below outlines complications of MPS VI by organ system. The clinical manifestations associated with MPS VI are heterogeneous; however, all patients will experience disease progression.

Patients with MPS VI express markedly lower growth rates than unaffected age-adjusted peers. The deviation in growth experienced by patients with MPS VI may be a sign of secondary problems and should prompt clinical surveillance for issues such as malnutrition, endocrine abnormalities or psychosocial deprivation.

MPS VI is a clinically heterogeneous condition with a spectrum of phenotypes that have variable onset and rates of progression.2 The figure below demonstrates the wide variability between a rapidly progressing patient and a slowly progressing patient. The rapidly progressing patient displays many of the phenotypic hallmarks of MPS VI.
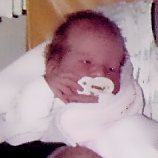
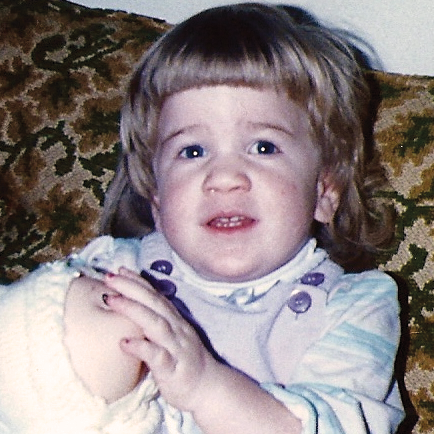


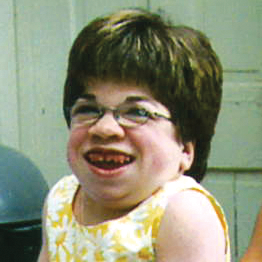
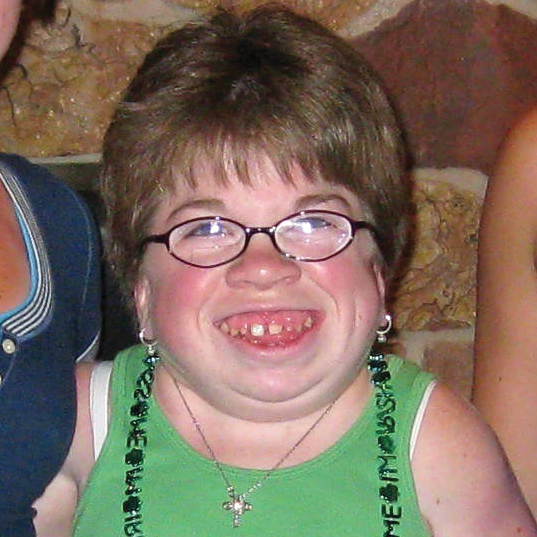
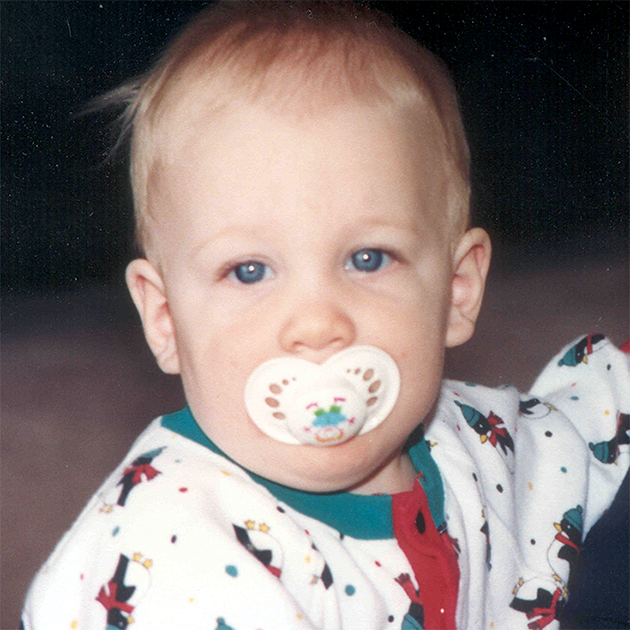

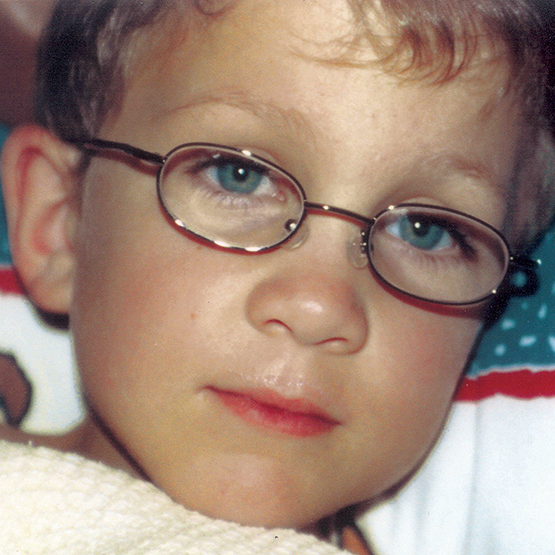



Differentiation into rapidly and slowly progressing disease is only apparent in extreme cases. If left untreated, disease progression will inhibit physical and functional well-being and results in a markedly shortened life span.4
Regardless of phenotype, affected individuals progress over the course of years and ultimately experience4
Patients with MPS VI usually die from infections, complications secondary to surgery or cardiopulmonary disease.4
MPS VI is an autosomal recessive condition caused by a deficiency of the enzyme N-acetylgalactosamine 4-sulfatase (also known as ASB). This deficiency results in the accumulation of the GAG dermatan sulfate in the lysosomes of a wide range of tissues.13,14

No specific ethnic group has been associated with an increased risk of MPS VI; however, some increased frequencies of specific mutations have been reported. There is a common mutation (1533del23) found in 23% of alleles in Brazilian patients with MPS VI. Additionally, one study demonstrated a high birth prevalence of MPS VI in the Turkish population living in Germany, as compared with the non-Turkish German population.2
Over the past decade, the management of MPS VI has evolved along with clinicians’ knowledge about the disease, such as its phenotypic variability and progression. Other factors, such as patients living into adulthood, highlight new areas for research, understanding and promise in management.
MPS VI affects multiple body systems, making management of a diverse spectrum of disease manifestations an important part of providing integrated care. Management should include use of adaptive or supportive devices, physical and occupational therapy, symptom-based medications, surgical interventions, and treatment to provide the deficient enzyme.4
Published in 2007, the “Management Guidelines for Mucopolysaccharidosis” provides recommendations across specialties for the management of patients with MPS VI. It recommends enzyme replacement therapy (ERT) as a treatment option to be considered in patients with MPS VI.4
In addition to initiating ERT where appropriate, the guidelines recommend system-specific management strategies for symptoms associated with GAG buildup.4
Lifetime management and procedural care from a multidisciplinary coordinated care team are critical components to optimising patient outcomes.16 Patients with MPS VI require early and regular assessments to evaluate disease progression across multiple organ systems and to detect and treat potential complications of the disease.4 The table below shows the schedule of assessments for patients with MPS VI recommended by the 2007 guidelines.

Patients with MPS VI often require surgical intervention to address the multisystemic complications of the disease. This surgical care is complicated by the nature of the disease.16
Patients with MPS VI suffer from multiple factors that can dramatically increase surgical risk and the need for monitoring, including the following16:
These factors complicate surgical and anaesthetic care, require pre-planning, and necessitate disease-specific techniques to increase optimal outcomes.17
Specialised perioperative procedures during anaesthesia, such as intubation and extubation, and use of an intraoperative neuromonitoring checklist, are essential to successful surgical interventions. An integrated surgical team consisting of MPS VI specialists is critical for positive, durable outcomes.16,17
As patients with MPS VI reach adulthood, their relationship with their medical team will change. To help manage this transition, individual plans are necessary to minimise treatment interruptions, extend support beyond the scope of paediatric care and parental support, and ensure that adult patients are knowledgeable in managing MPS VI.18
These transition plans should be tailored to each patient’s specific needs, so that those who can take over their own care will have the necessary tools, and those who are more limited will have the appropriate care and services in place to support them. The plans should include an assessment to determine the patient’s capacity to achieve his or her outlined goals, as well as his or her knowledge and ability to communicate information about his or her condition.18
References: 1. Thümler A, Miebach E, Lampe C, et al. Clinical characteristics of adults with slowly progressing mucopolysaccharidosis VI: a case series. J Inherit Metab Dis. 2012;35(6):1071-1079. doi:10.1007/s10545-012-9474-1. 2. Valayannopoulos V, Nicely H, Harmatz P, Turbeville S. Mucopolysaccharidosis VI. Orphanet J Rare Dis. 2010;5:5. doi:10.1186/1750-1172-5-5. 3. Swiedler SJ, Beck M, Bajbouj M, et al. Threshold effect of urinary glycosaminoglycans and the walk test as indicators of disease progression in a survey of subjects with mucopolysaccharidosis VI (Maroteaux–Lamy syndrome). Am J Med Genet A. 2005;134A(2):144-150. doi:10.1002/ajmg.a.30579. 4. Giugliani R, Harmatz P, Wraith JE. Management guidelines for mucopolysaccharidosis VI. Pediatrics. 2007;120:405-418. doi:10.1542/peds.2006-2184. 5. Muhlebach MS, Wooten W, Muenzer J. Respiratory manifestations in mucopolysaccharidoses. Paediatr Respir Rev. 2011;12(2):133-138. doi:10.1016/j.prrv.2010.10.005. 6. Lin H-Y, Chen M-R, Lin C-C, et al. Polysomnographic characteristics in patients with mucopolysaccharidoses. Pediatr Pulmonol. 2010;45(12):1205-1212. doi:10.1002/ppul.21309. 7. Kampmann C, Lampe C, Whybra-Trumpler C, et al. Mucopolysaccharidosis VI: cardiac involvement and the impact of enzyme replacement therapy. J Inherit Metab Dis. 2014;37(2):269-276. doi:10.1007/s10545-013-9649-4. 8. Willoughby CE, Ponzin D, Ferrari S, Lobo A, Landau K, Omidi Y. Anatomy and physiology of the human eye: effects of mucopolysaccharidoses disease on structure and function—a review. Clin Exper Ophthalmol. 2010;38(suppl 1):2-11. doi:10.1111/j.1442-9071.2010.02363.x. 9. Ganesh A, Bruwer Z, Al-Thihli K. An update on ocular involvement in mucopolysaccharidoses. Curr Opin Ophthalmol. 2013;24(5):379-388. doi:10.1097/ICU.0b013e3283644ea1. 10. Kantaputra PN, Kayserili H, Güven Y, et al. Oral manifestations of 17 patients affected with mucopolysaccharidosis type VI. J Inherit Metab Dis. 2014;37(2):263-268. doi:10.1007/s10545-013-9645-8. 11. Quartel A, Hendriksz CJ, Parini R, Graham S, Lin P, Harmatz P. Growth charts for individuals with mucopolysaccharidosis VI (Maroteaux-Lamy Syndrome). JIMD. 2015;18:1-11. doi:10.1007/8904_2014_333. 12. Data on file. BioMarin Pharmaceutical Inc. 13. NAGLAZYME [package insert]. Novato, CA: BioMarin Pharmaceutical Inc; 2013. 14. Giugliani R, Lampe C, Guffon N, et al. Natural history and galsulfase treatment in mucopolysaccharidosis VI (MPS VI, Maroteaux-Lamy syndrome)—10-year follow-up of patients who previously participated in an MPS VI Survey Study. Am J Med Genet A. 2014;164A(8):1953-1964. doi:10.1002/ajmg.a.36584. 15. Kakkis ED. Enzyme replacement therapy for the mucopolysaccharide storage disorders. Expert Opin Investig Drugs. 2002;11(5):675-685. 16. Walker R, Belani KG, Braunlin EA, et al. Anaesthesia and airway management in mucopolysaccharidosis. J Inherit Metab Dis. 2013;36(2):211-219. doi:10.1007/s10545-012-9563-1. 17. Vitale MG, Skaggs DL, Pace GI, et al. Delphi Consensus Report: Best practices in intraoperative neuromonitoring in spine deformity surgery: development of an intraoperative checklist to optimize response. Spine Deformity. 2014;2(5):333-339. doi:10.1016/j.jspd.2014.05.003. 18. American Academy of Pediatrics, American Academy of Family Physicians, American College of Physicians, Transitions Clinical Report Authoring Group, Cooley WC, Sagerman PJ. Supporting the health care transition from adolescence to adulthood in the medical home. Pediatrics. 2011;128(1):182-200. doi:10.1542/peds.2011-0969.
